| Home
| IdeaBook
| Orders
| Price/Applic
| Price/Vendor
| Protocols|
Protein Purification: Column Chromatography - Ion exchange; Dialysis and Concentration
BCH5425 Molecular Biology and Biotechnology
BCH5425 Molecular Biology and Biotechnology
Spring 1998
Dr. Michael Blaber blaber@sb.fsu.edu Florida State University, adapted with permission.
Lecture 30
Column chromatography
After initial fractionation steps the typical procedure is to move to column chromatography.
- In column chromatography we have a glass tube (column) which is filled with a material ("resin") which has certain physical/chemical characteristics.
- These characteristics allow it to interact in various ways with different proteins.
- Some common types of chromatographic resins include:
- Ion exchange
- Hydrophobic
- Gel filtration
- Affinity
Ion exchange
Ion exchange resins contain charged groups.
- These may be acidic in nature (in which case the resin is a cation exchanger)
- or basic (in which case it is an anion exchanger).
- Cation and anion exchangers may be broken down further into weak and strong exchangers (reflecting binding affinity).
|
Type of exchanger |
Functional group |
Common name |
|
Weak cation exchanger |
carboxymethyl |
CM cellulose/sephadex |
|
Strong cation exchanger |
sulfopropyl |
SP sephadex |
|
Weak anion exchanger |
diethylaminoethyl |
DE cellulose/sephadex |
|
Strong anion exchanger |
quaternary amine |
QAE sephadex |
- Usually, samples are loaded under low ionic strength conditions and bound material is eluted using either a step or gradient elution of buffer with higher ionic strength.
- Generally speaking, a protein will bind to a cation exchange resin if the buffer pH is lower than the isoelectric point (pI) of the protein, and will bind to an anion exchange resin if the pH is higher than the pI.

- Knowledge of the pI of the protein is helpful in designing a purification protocol using ion exchange resins. To have complete ionization one needs to have the mobile phase pH, two pH units away from the nearest pKa or pKI. If you can adjust the pH of the binding and/or elution buffers two pH units from the pI of your protein, then the column's ionic functonal groups will have distinct ionic species to separate, since they will be either totally ionized or total protonated under these conditions. Then one only needs to compare the pH of the mobile phase, the pKa of the column and the pKI of the protein to determine if a pH gradient or a salt gradient will most easily elute the protein from its impurities.
When I was learning to do IEX of proteins, I would draw a pH nomograph for both the protein (relative to a neutral pH buffer) and the IEX functional group choices of the column, to determine which would be most useful:
pKI acidic (neg. chg. carboxyl, COO-, predominates) ------ protein ------ pKI basic (pos. chg. amine, NH3+, predominates)
SCX (SO4, pKa ~1.5) WCX (COOH, pKa 3.8-4.2)) ------ column ------ WAX (DEAE pKa ~8.0) SAX (QAE, pKa ~12.0)
-
One can get elution by either increasing the amount of salt and/or by changing the pH to either remove the charge from the protein or from the column (for weak ion exchange columns). The closer you are to the pKa of the column or pKI of the protein the less salt is required, but it also means that the protein is potentially in more than one charge state, and thus this isn't a good method. One pH unit away is 90% ionized while two pH units is 99% ionized.
If you don't know the pI of the protein, use a pH of 5.5 - 6.5 for either type of weak ion exchange column and run a salt gradient to see what results. Then modulate the pH by running an acid buffer gradient (acetic or formic acid for volatile buffers, or a phosphate, non-volatile buffer) or, if the column is pH stable, a basic buffer gradient (ammonium bicarbonate or 0.1% ammonia, for volatile buffers, or TRIS, etc. for non-volatile ones). Strong ion exchangers cannot have their charge completely removed, so if using one of these resins for proteins, you will remove the charge from the protein before that of the column and it will elute on this basis instead. Knowing whether the pKI of the protein is above or below the pKa of the column (from the nomograph), will tell you which pH buffer to use to most easily effect elution.
Generally speaking, ion exchange columns are short and fat in dimensions.
Elution of proteins from ion exchange resins
- Proteins bound to ion exchange resins are bound via non-covalent ionic (salt-bridge) interactions. We can compete for these ionic binding sites on the resin with other ionic groups, namely, salts.
- There are two general types of methods when eluting with a salt solution: 1. Gradient elution and 2. Step elution
- A gradient elution refers to a smooth transition of salt concentration (from low to high) in the elution buffer. Weakly binding proteins elute first, and stronger binding proteins elute last (i.e. they require higher salt concentrations in the buffer to compete them off the column)
- A gradient salt concentration can be made using a gradient maker. In its simplest form, this consists of two containers (must be the same shape) connected by a siphon (or tube at the bottom). One container contains the low salt buffer, and the other contains high salt buffer. The buffer is withdrawn from the low salt container:

- This will produce a linear gradient from low to high salt concentrations over the total volume of the gradient

- If we know the concentration range of salt over which a protein of interest will elute we can simply elute with a buffer containing that concentration of salt. This is known as a step elution.
- Step elutions are generally faster to run, and elute the protein in a smaller overall volume than with gradient elutions. They generally work best when contaminants elute at a significantly different salt concentration than the protein of interest

Note that after ion exchange chromatography the protein of interest will be in a buffer with a potentially high salt concentration. This must be taken into account before proceeding with the next step in the purification scheme
- Dialysis
- After an ammonium sulfate precipitation step, or an ion exchange chromatography step, the protein of interest may be in a high salt buffer. This may be undesirable for several reasons. How do we get rid of salt in our sample?
- One of the most common methods is that of dialysis
- The method of dialysis makes use of semi-permeable membranes. In the simplest example, this membrane is manufactured in the form of tubing (looking much like a sausage casing)
- The main feature of this membrane is that it is porous. However, the pore size is such that while small salt ions can freely pass through the membrane, larger protein molecules cannot (i.e. they are retained). Thus, dialysis membranes are characterized by the molecular mass of the smallest typical globular protein which it will retain.
- This is commonly referred to as the cutoff of the tubing (e.g. Spectrapore #6 dialysis tubing has a cutoff of 1,000 Daltons, meaning that a 1,000 Dalton protein will be retained by the tubing but that smaller molecular mass solutes will pass through the tubing)
- Dialysis proceeds by placing a high salt sample in dialysis tubing (i.e. the dialysis "bag") and putting it into the desired low salt buffer:

- Over time the concentration of low molecular mass solutes within the bag, and in the low salt buffer, will come achieve equilibrium. In practical terms (for the above case) salt molecules will diffuse out of the bag into the low salt buffer:

- At equilibrium the salt concentration of the sample can be calculated as follows:

Note: often the buffer salt concentration is 0 M
- The buffer volume for the dialysis is a function of the required final concentration of salt in the sample
Dialysis example
We have a 10ml protein sample from an ion exchange column elution pool which contains 1.0M NaCl. For our next step in the purification we can have no more than 1mM NaCl in the sample.

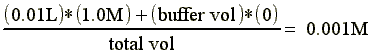

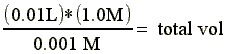
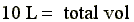
Therefore, the required buffer volume would be (total vol - sample vol) = 9.990 L (or ~ 10 L)
- Thus, if we dialyzed 10mls of sample (with 1.0M NaCl conc) in 10 L of water after equilibrium the NaCl concentration in the sample would be 1.0 mM.
- Note that in the above example this would commonly be referred to as a "1:1,000" dialysis.
- Suppose that we don't want to make up 10 L of buffer? We can actually achieve the same results with two sequential "1:32" dialyses (i.e. the square root of the 1:1,000 dialysis - in other words, two sequential 1:32 dialyses is equivalent to a single 1:1,000 dialysis):
First dialysis versus 310 ml of buffer: sample NaCl conc will be (10*1.0)/(320) = 31 mM
Second dialysis versus 310 ml of buffer: sample NaCl conc will be (10*0.031)/(320) = 0.97 mM
Thus, instead of making 10 L of buffer, we could make only 620 ml and achieve the same results with two dialysis steps
- In this case, removing the salt would take twice as long, i.e. we need to perform two dialysis steps. How long does dialysis take?
A useful rule of thumb is that for most types of dialysis tubing the dialysis is 80% compete after four hours
- One consequence of dialysis to watch out for is that while salt ions are moving out of the bag, water molecules are moving into the bag. Thus the volume of sample may actually increase (the bag will swell) and, therefore, the protein concentration will decrease
- In the extreme case, the bag may actually swell to the point of rupture. Therefore, it is a good idea not to fill the bag completely, but leave a void to allow for potential swelling.
Concentration
- What if our protein sample is too dilute for our needs? How can we concentrate our samples?
- One common method is, again, to use a semi-permeable membrane for this purpose.
- A very simple method is to place our sample in a dialysis bag and coat it with a high molecular weight solute which can readily be dissolved by the buffer.
- For example, polyethylene glycols and polyvinyl pyrolidones can have very large molecular masses (i.e. 20,000 Da). These compounds are also readily dissolved in water. If our sample in a dialysis bag is coated with dry forms of the above polymers, water will leave the dialysis bag (it can go through the pores) and hydrate the polymers. The result is a decrease in volume of buffer in the dialysis bag (the protein will be concentrated).
- In another variation, the semi-permeable membrane is manufactured into a flat disk and placed at the bottom of a container which holds our sample. In one method the container is pressurized and forces buffer out of the container (protein is retained and is concentrated). In another method, the vessel is centrifuged and the centripetal force achieves the same goal as pressure in the previous example.
For both dialysis and concentration, it is essential that the membrane does not interact with the protein (i.e. has no affinity for, and will not bind, the protein)
- With the pressure type concentrators, dialysis and concentration can be achieved in tandem. For example, the sample can be concentrated and then buffer added to the sample. The sample is then concentrated again. Every time buffer is added the salt concentration is reduced. After repeated cycles of this, the salt concentration is at the desired level and the sample is concentrated to the desired final volume.
1998 Dr. Michael Blaber
Copyright© 1995-2024 The Nest Group, Inc.™ All rights reserved (established 1984)
For more information contact:
The Nest Group, Inc.™ 17 Hayward St., Ipswich, MA 01938-2041 USA
Please e-mail to
webmaster.
| Home
| IdeaBook
| Ordering
| Price/Applic
| Price/Vendor
| Protocols|
If you have problems or comments concerning
our WWW service, please send an e-mail to
webmaster.
About Us | GDPR Privacy Policy | Trademarks | Contact Us
Last Updated: 08/20/21
